









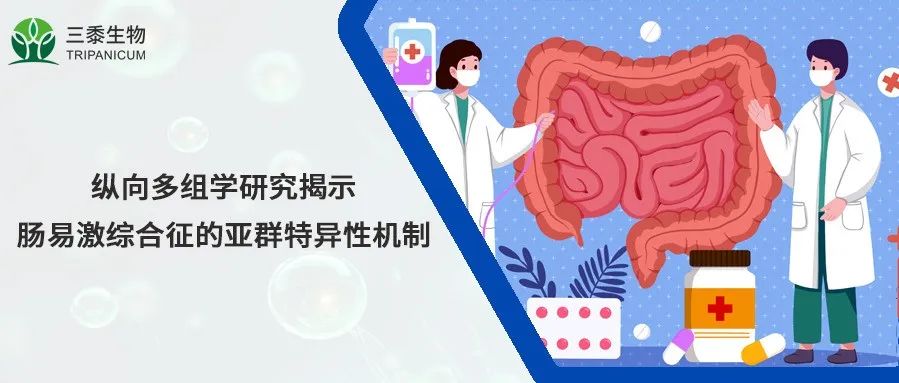
腸易激綜合征(IBS)全球高發,以反復發作的腹痛或不適為特征,分便秘型、腹瀉型等亞型,其發病機制與胃腸動力、腸分泌、內臟高敏感性和腸通透性等改變相關,腸道微生物群、飲食、遺傳等因素可對其產生影響。雖然移植實驗證實了腸道微生物群在 IBS 中的作用,但因缺乏理想動物模型模擬IBS病理生理學完整特征,需開展人類研究。現有人類研究受橫斷面采樣和亞型分層不足等條件限制,造成微生物組研究結果不一致,且 IBS 癥狀具周期性,橫斷面樣本無法體現疾病時間變異性,加之人類與動物宿主生理學差異,阻礙了對腸道微生物群在 IBS 中機制的理解。因此,這篇刊登在Cell上的研究《Longitudinal Multi-omics Reveals Subset-Specific Mechanisms Underlying Irritable Bowel Syndrome》對 IBS 患者亞群進行縱向多組學研究,以探究由微生物代謝改變引起的IBS亞型特異性機制。

研究對象:腸易激綜合征患者及健康人群的糞便、腸道活檢樣本,無菌小鼠,培養菌株
技術方法:
宏基因組,代謝組,轉錄組,Ussing chamber實驗,小鼠菌株移植
技術路線:
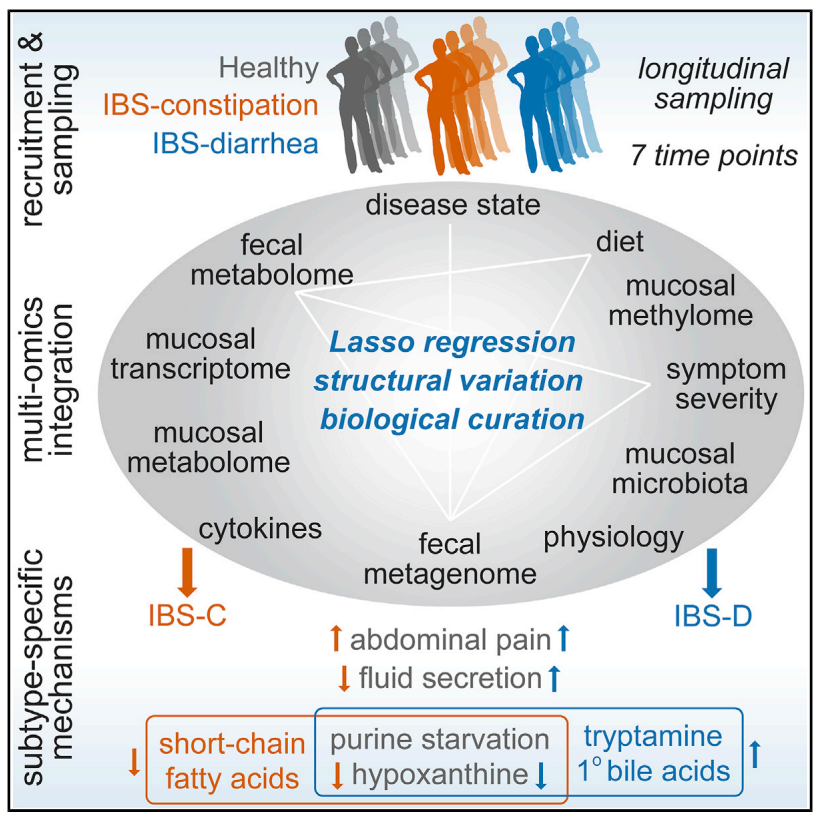
1.縱向采樣克服了橫斷面微生物組研究中的異質性
通過對比健康對照(HC)與 IBS 患者的縱向微生物組數據,發現單個時間點的菌群豐度差異不一致,與平均數據中的變化不重合,且數據顯示個體間變異大于個體內變異。而平均數據能可靠揭示亞型特異性變化:便秘型腸易激綜合征(IBS-C) 和腹瀉型腸易激綜合征患者(IBS-D)患者的鏈球菌屬豐度均顯著高于健康人群,IBS-D 患者的 Synergistetes 門豐度則顯著低于HC。PCoA 分析顯示,IBS 患者糞便菌群組成按亞型聚類,IBS-C 、 IBS-D 的菌群分散度與健康組減互相都存在顯著差異(圖 1D和1E)。進一步分析發現,IBS-C 患者的菌群不規則性(BCDI 評分)顯著高于 IBS-D 和健康組,約 31.7% 的 IBS-C 樣本數據位于健康組菌群分布的第 90 百分位數,高于IBS-D(14.1%)(圖 1F),提示其菌群結構異常更為突出。這些結果表明,縱向采樣結合數據平均化處理能更精準地捕捉 IBS 相關的菌群動態變化,避免橫斷面采樣遺漏造成的誤差,為揭示疾病亞型機制提供了可靠方法學支持。
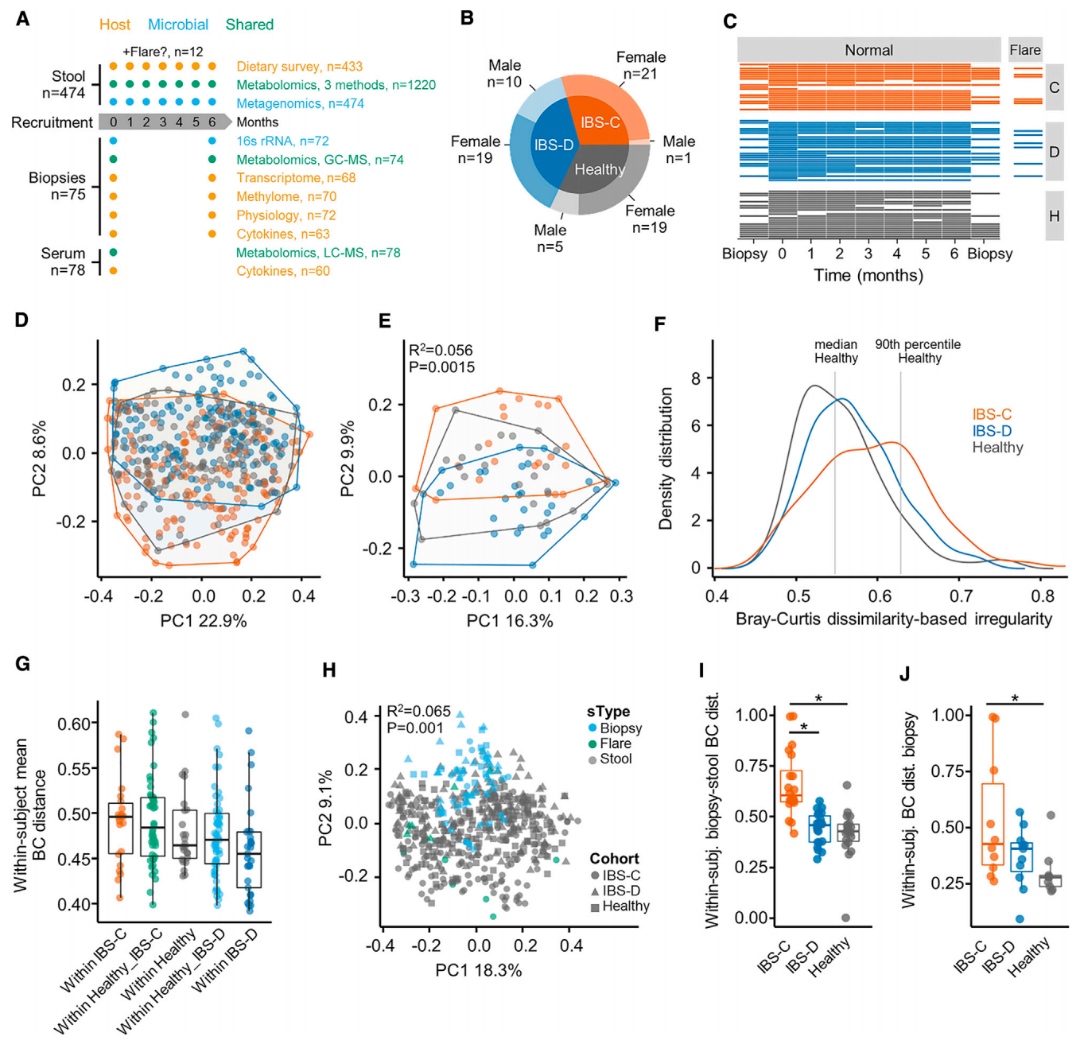
圖 1. 便秘型腸易激綜合征(IBS-C)患者的腸道菌群組成更具特異性且變異性更高
2.縱向采樣揭示 IBS-C 微生物群隨時間的變異性更高
通過分析縱向采樣數據發現,IBS-C患者的糞便微生物群組成隨時間變異性顯著高于健康對照和IBS-D,平均香農 α多樣性較 IBS-D 高(p=0.016)(圖1G)。進一步分析顯示,黏膜與管腔的微生物組成存在顯著差異:IBS 患者的黏膜相關微生物群中變形菌門水平升高,而 IBS-C 患者的黏膜菌群與管腔菌群的相似度在三組樣本中最低,這可能與其腸道轉運時間延長導致菌群分化有關(圖1H和1I)。此外,IBS-C患者的黏膜微生物群在不同時間表現出的個體內變異性也高于其他組(圖1J)。這些發現表明,IBS-C 的腸道微生物群在組成多樣性、空間分布及時間動態上均存在獨特異常。
3.腸易激綜合征癥狀嚴重程度與腸道微生物群功能變化相關
綜合腹痛強度、頻率、腹脹、對排便習慣的不滿以及 IBS 對生活的總體影響等因素,對IBS癥狀在各特定采樣點的嚴重程度進行評分,研究人員發現重度IBS-D患者樣本中有20多種乳桿菌屬的相對豐度高于輕中度患者,且與受試攝入益生菌或乳制品無關。糞便宏基因組數據進行KEGG Orthology數據庫功能分析,結果顯示重度便秘型IBS(IBS-C)和 IBS-D 分別有74個和44個KO項異常,其中2種亞型的重度患者樣本中均存在酒精脫氫酶(ADH)相關KO項(區別于輕中度IBS)。盡管ADH酶遺傳多樣性較高,豐度難以與特定的代謝產物直接關聯,但根據兩類IBS亞型的共同主要癥狀都是腹痛,研究者推測其活性很可能與這一癥狀有關。布里斯托爾糞便量表記錄也顯示的糞便形態及排便前腹痛與特定細菌和代謝物相關。
4.代謝組學與生理測量相結合為腸道微生物群代謝對胃腸功能的影響提供了機制性見解
研究者對反映管腔和黏膜相關樣本生化特征的微生物組代謝進行了量化分析。1H - 核磁共振(NMR)光譜顯示,IBS-C 患者糞便中的丙酸、丁酸和乙酸等短鏈脂肪酸(SCFAs)水平較健康對照組顯著降低,結腸黏膜樣本中的乙酸也顯著減少,且該變化與膳食纖維攝入量無關(圖 2A、2B)。尤斯灌流室實驗顯示,IBS-C 患者結腸上皮對血清素(5-HT)的分短路電流(isc)泌反應較健康樣本減弱(圖 2C),印證了SCFAs 缺乏導致腸道分泌減少、糞便含水量降低的病理機制。
對于 IBS-D 患者,研究發現其糞便中色氨酸及代謝產物色胺水平顯著升高(圖2D),而小鼠實驗顯示細菌衍生的單胺色胺可通過激活 5-HT?R 受體促進腸道分泌。通過尤斯灌流室實驗發現,IBS患者和健康對照組樣本中缺乏色胺誘導isc變化的顯著差異。進一步結合人體中初級膽汁酸(BAs)和鵝去氧膽酸(CDCA)等物質相關的增加腸道液體分泌通路, LC-MS/MS 檢測發現IBS-D患者中的未結合初級BAs含量顯著低于HC組,IBS-C患者樣本則相反(圖 2E)。尤斯灌流實驗證實了CDCA 誘導的Isc顯著增加,提高IBS-D患者糞便含水量的作用。結腸活檢樣本isc差異顯示,IBS-D患者的基線isc顯著高于其他組別(圖 2F)。證實了初級BA水平差異和色胺綜合影響了腸道分泌,通過調控腸道上皮分泌參與疾病表型形成。

圖 2. 代謝組學與生理功能檢測的整合為腸道微生物群代謝對胃腸功能的影響提供機制性見解
5.整合微生物組 - 代謝組分析鑒定出 IBS 中一條新的微生物代謝途徑
非靶向代謝組學和潛在結構投影判別分析(PLS-DA)顯示,IBS-C 患者糞便中的賴氨酸、尿嘧啶和次黃嘌呤水平較HC組均顯著降低(圖 3A-3C),IBS-D 患者的次黃嘌呤也較低,但顯著水平低于IBS-C患者。次黃嘌呤作為腸上皮細胞的能量來源,其水平降低可能影響腸道屏障發育和修復,其在IBS患者糞便中水平的降低可能反應了腸道微生物組的影響。
宏基因組功能分析表明,IBS-C 患者糞便中黃嘌呤磷酸核糖基轉移酶(XPRT)和黃嘌呤脫氫酶 / 氧化酶(XO)模塊豐度較HC組顯著升高(圖 3D),提示腸道微生物群的嘌呤分解代謝增強。進一步分析發現,IBS-C 患者糞便中與能量代謝相關的 TCA 循環模塊(如 L - 乳酸脫氫酶、丙酮酸脫氫酶)及替代呼吸途徑相關模塊(亞硫酸鹽還原酶 / 鐵氧還蛋白、亞硫酸鹽、亞硝酸鹽還原酶和細胞色素 C 氧化酶)豐度也顯著增加(q<0.1,圖 3D),同時超氧化物還原酶模塊升高。這些結果表明IBS患者的微生物組的次黃嘌呤李永和分解能力增強,從而影響了患者的腸道屏障功能,為 IBS 的病理機制提供了新的代謝視角。

圖 3. 微生物組 - 代謝組整合分析鑒定出腸易激綜合征(IBS)中一條新的微生物代謝通路
6.微生物基因區域分析證實與膽汁酸、丁酸和次黃嘌呤代謝的關聯
基于線性模型的直接多變量相關分析(Maaslin)發現,健康對照、IBS-C 和IBS-D 分別存在60、28 和46 個顯著的代謝物-物種相關性,其中HC分別與IBS-C或IBS-D有12個共同存在的相關性關系,兩種IBS亞型間存在2個,沒有同時存在于3組的相關性關系。
進一步通過結構可變基因組區域與代謝物豐度相關聯(SV關聯)分析可能導致組間代謝物輸出差異的特定細菌基因組區域,從微生物組中鑒定出了16個完全缺失區域(DRs)和20個可變豐度區域(VRs),分別與9種和8種代謝物相關,其中包括CDCA、丁酸和次黃嘌呤等代謝物(圖 4A–4B)。

圖 4. 基于微生物基因區域關聯分析提示特定腸道微生物成員對次黃嘌呤的消耗作用
7.微生物代謝對管腔次黃嘌呤水平的貢獻
基于基因區域分析結果,研究者選取了2株與此前鑒定出Lachnospiraceae sp. 3_1_46FAA 基因組相似的毛螺菌科菌株,聯合陽性對照菌 Hungatella hathewayi(含 XO 基因)和陰性對照菌長雙歧桿菌(不編碼XO基因)進行體外培養,LC-MS 檢測顯示前兩者的培養基中次黃嘌呤水平顯著降低(圖4C)。無菌小鼠體內實驗進一步發現,定植毛螺菌科 sp. 2_1_58FAA 的無菌小鼠在使用飲用水補充次黃嘌呤的條件下,盲腸內容物中次黃嘌呤水平仍較定植長雙歧桿菌的小鼠顯著下降(圖4D-E)。該結果印證了微生物可通過 XO 等相關酶促反應消耗次黃嘌呤,且體內次黃嘌呤水平由微生物的生產與消耗代謝平衡共同決定。
8.腸易激綜合征患者癥狀發作的腸道微生物群和微生物代謝物改變
IBS的癥狀嚴重程度隨時間變化。為證實IBS癥狀加重的腸道微生物基礎,研究人員對自我報告發生癥狀惡化的樣本進行了檢查。
與非發作期IBS樣本相比,發作期糞便樣本的 BCDI(基于 Bray-Curtis 相異度的不規則性評分)顯著升高(圖5A-B),香農α多樣性降低(圖5C),菌群結構穩定性下降。微生物分類群分析顯示,168 種細菌與 IBS 整體發作相關,其中IBS-C和IBS-D分別有40 種和7 種特征菌,且除古菌Halobiforma nitratireducens豐度升高外(圖 5D),其余相關菌屬普遍減少。代謝物檢測發現,IBS-C和IBS-D 發作樣本中初級 BA 水平均顯著升高(圖5E-F),可能參與腹痛癥狀的產生。功能宏基因組KO模塊分析顯示,IBS-D 發作期的酒精脫氫酶和黃嘌呤氧化酶(XO)模塊豐度增加,與三羧酸循環和呼吸相關通路激活一致。個體水平的時間進程分析進一步表明,IBS-C 患者發作前BCDI評分隨時間上升,且 6/11 的患者在發作時膽汁酸、4/11 的患者色胺等分泌性代謝物升高。這些發現證實,不同患者可能存在獨特的IBS 癥狀加重微生物-代謝驅動模式。

圖 5. 腸道微生物群及代謝物的改變是腸易激綜合征(IBS)患者癥狀發作的基礎
9.微生物組和代謝組數據與轉錄組和表觀遺傳差異的整合揭示了 IBS 中新型宿主-微生物相互作用
轉錄組分析顯示,IBS-C和IBS-D分別有82和78 個差異表達基因與HC組有差異,其中17個基因重疊。KEGG通路富集分析顯示,IBS 患者中免疫和炎癥相關通路富集,包括IBS-C中調節平滑肌收縮相關基因(PTGDS)、B細胞抗原反應基因(CD19 和CD22)抗原呈遞基因(HLA-DQA1和HLA-DQB1)基因。
表觀基因組分析鑒定出和HC組相比,IBS-C組有54個差異甲基化區域(DMR),IBS-D有75個,IBS亞型間比較有39個。其中KCNE4和AQP1基因甲基化異常,其低表達可能導致腸道分泌功能障礙。進一步對DMR基因進行KEGG通路富集分析,發現抗原加工和呈遞通路在兩種IBS亞型種均有富集,與人類白細胞抗原(HLA)II 類基因有關。與 HC 活檢相比,IBS-C 活檢組織中表現出編碼HLAII 類分子相關基因表達升高4倍,其中的HLA-DQA1與普通擬桿菌存在相關性,提示了細菌抗原與IBS-C之間的潛在作用。
通過 Lasso 回歸模型構建的跨組學網絡顯示,乙酸鹽與 PGLYRP1、KIFC3 基因形成調控樞紐(圖 6B)。其中 PGLYRP1 與革蘭氏陽性菌 Peptostreptococcaceae 負相關,可能通過抑制肽聚糖合成產生殺菌活性;KIFC3 則與腸道屏障功能相關。這些發現通過多組學整合揭示了 IBS 中 “微生物代謝-宿主基因表達-免疫調控” 的級聯效應,為解析 IBS 亞型特異性機制提供了從基因表達到微生物互作的多層次證據(圖 6)。

圖 6. 基于 Lasso回歸的多組學整合分析結果
10.多組學整合鑒定出結腸上皮嘌呤饑餓是腸易激綜合征的潛在新機制
IBS-C和IBS-D患者糞便中次黃嘌呤水平顯著降低(圖 3和7),人黃嘌呤氧化酶表明次黃嘌呤庫的消耗可能是微生物和宿主 XO 活性增加的結果。
由于腸上皮主要依賴嘌呤補救途徑合成腺苷酸(圖 7C),該途徑的重要組成部分——嘌呤核苷磷酸化酶(PNP)基因在IBS患者樣本中表達量上調 2 倍(圖 7A),與次黃嘌呤水平呈負相關(圖 7D),且 Illumina 全局篩選陣列顯示與宿主遺傳學變異無關。
這些發現共同提出模型:微生物和宿主嘌呤核苷酸降解升高誘導結腸組織代謝應激,進而可能通過增加嘌呤補救導致代償反應。嘌呤核苷酸水平低可能導致上皮能量狀態和黏膜修復能力降低,這可能部分構成 IBS 的病理生理學基礎。

圖 7. IBS 的多組學整合視角揭示嘌呤補救通路中的微生物-宿主互作機制
本研究通過縱向多組學技術(宏基因組、代謝組、轉錄組及甲基組)分析,揭示腸易激綜合征(IBS)的亞型特異性機制。IBS-C 患者腸道菌群多樣性高且不穩定,短鏈脂肪酸(SCFAs)水平降低,導致腸道分泌功能減弱;IBS-D 患者則表現為色胺和初級膽汁酸升高,促進腸道分泌。此外,IBS 患者普遍存在嘌呤代謝異常,次黃嘌呤水平降低,伴隨宿主嘌呤補救途徑基因(PNP、XDH)表達上調,提示結腸上皮 “嘌呤饑餓” 可能是共同病理基礎。研究強調縱向采樣和多組學整合的重要性,為 IBS 的機制分型和靶向治療(如調控嘌呤代謝、SCFAs 補充)提供新思路。
滑動查看更多:


排版:野凌
審核:三黍生物企宣部

